1 About UbiBrowser
The ubiquitination mediated by E1-E2-E3 cascade is crucial to protein degradation, transcriptional regulation and cellular signaling in eukaryotic cells. The high specificity of ubiquitination is regulated by the interaction between E3 ubiquitin ligases and their target substrates. UbiBrowser is an integrated bioinformatics platform to predict and present the proteome-wide human E3-substrate network based on naïve Bayesian network. It currently contains 1295 literature reported E3-substrat interactions and 8,255 predicted E3-substrate interactions. Totally 20220 human proteins’ corresponding E3 ligases can be explored. Furthermore, 4560 human proteins’ 19, 451 ubiquitination sites can also be explored.
2 Main functions
UbiBrowser is designed to allow users to explorer the predicted and reported E3-substrate interactions for a query E3 or substrate, and ubiquitination sites for substrate protein. Three major views are offered by UbiBrowser: “network view of the predicted E3 ligase/substrate”, “table view of the literature reported E3 ligase/substrate” and “sequence view of substrate mediated by specific E3 ligase”.
“Network view of the predicted E3 ligase/substrate” shows the predicted E3-substrate interactions. There are two modes in this view: confidence mode and evidence mode. Confidence mode reflects the confidence of predicted E3-substrate interactions and evidence mode reflects the biological evidences supporting the prediction.
“Table view of the literature reported E3 ligase/substrate” present the E3-substrate interactions extracted from literatures.
“Sequence view of substrate mediated by specific E3 ligase” provides the known ubiquitination sites for query protein and predictive E3 recognizing domain/motif.
3 Browser requirements
UbiBrowser supports the latest versions of the Chrome, Firefox, Opera, Safari and Internet Explorer (version 10 and 11).
4 Data input
4.1 UbiBrowser supports three input types.
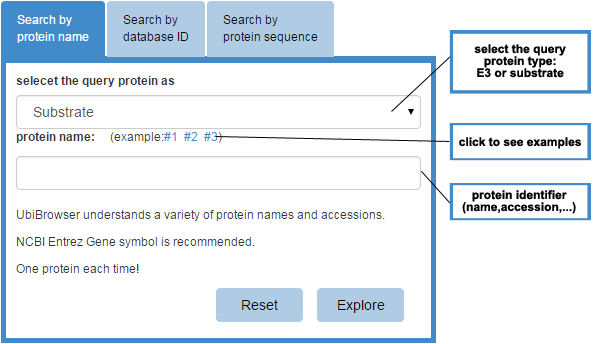
Figure 1 Input form
Please use the input form at the index page to query your protein. There are three tab controls in the input form and you can query your protein by entering protein name (gene symbol), protein accession number (UniprotAC/ID or GeneID) or protein sequence. You should select the query protein as E3 or substrate first, then you can submit your protein name/protein database ID/protein sequence in the under text field.
Attention:only one protein each time.
4.2 Specify the protein
When you search the protein by protein name or sequence, there might be multiple matches, and you should specify the query protein.
When you search the E3 or substrate protein by protein name, all the matched entries will be shown in a table with short descriptions to help you specify your query protein.
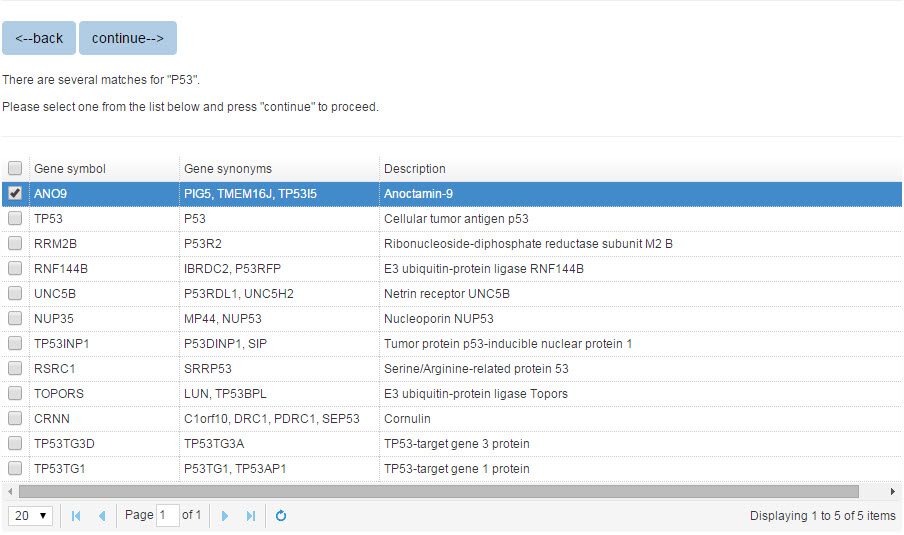
Figure 2. The list of candidate proteins
If you search the protein by protein sequence, the sequence identity score from BLAST will be listed in the parenthesis after the description.
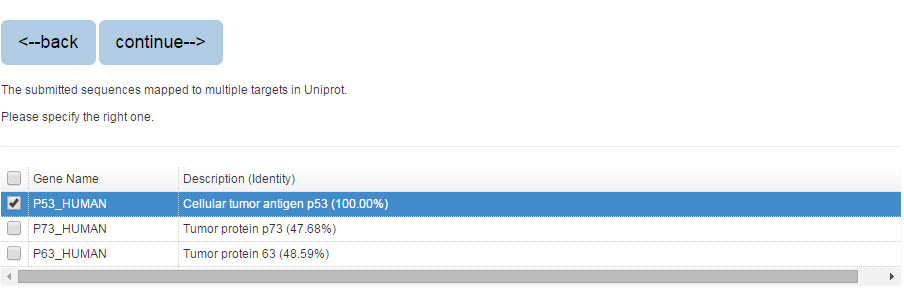
Figure 3. The list of blast result
Please specify the protein and click “continue” for result page.
5 Network view of the predicted E3 ligases/substrate.
Network view of the predicted E3 ligases/substrate is the default view. There are two modes in this view, confidence mode and evidence mode. You can press the mode exchange button to switch between these two modes. There are six function parts in this view. A: E3-substrate network. B: Statistics table. C: Filter. D: E3 hierarchical tree. E: Network picture export. F: Mode exchange button. G: Prediction summary.

Figure 4. Network view
5.1 Predicted E3-substrate network
This network shows the E3-substrae interactions for your query E3 ligase or substrate.
1) querying a substrate (left: confidence mode; right: evidence mode)
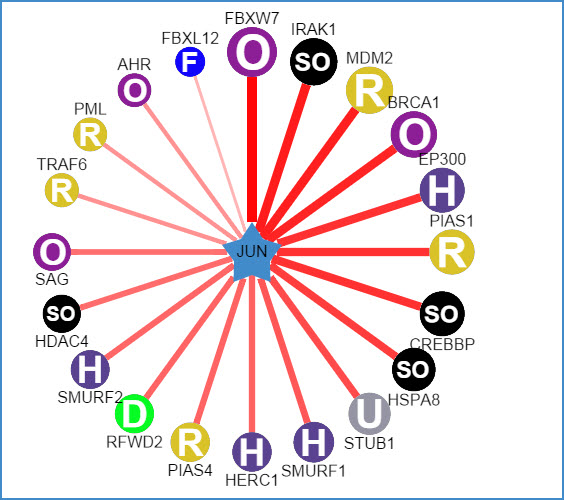

Figure 5. Network view for substrate proteins
Your query substrate locates in the center of the canvas. The predicted E3 ligases surround the substrate. The node colors and characters reflect the E3 type. The edge width, the node size and the edge shade are corrected with the confidence score. Node size, edge color depth, and edge width is proportional to the confidence score. Clicking surrounding nodes will link to the sequence view for the substrate andclicking the edges to the view of supporting evidences about these E3-substrate interactions.
2) querying an E3 ligase (left: confidence mode; right: evidence mode)
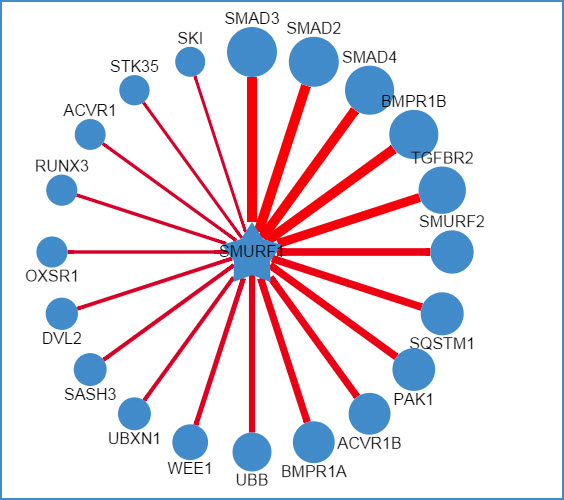
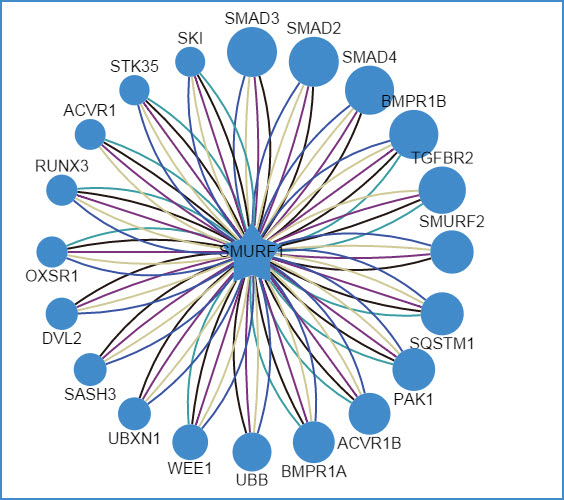
Figure 6. Network view for E3 ligases
Your query E3 ligase locates in the center of the canvas. The predicted substrates surround the E3 represented by the blue nodes.
Restricted to the canvas size, only top 20 predicted interactions are represented.
5.2 Confidence level
In order to give every predicted E3-substrate a confidence level, we calculated their “significance score” based on the following steps: we sort all the predicted E3-protein pairs by their confidence score in descending order and get each pair’s rank. Each prediction’s “significance score” is calculated as the ratio of its rank to the number of all the predicted pair multiplies by the substrates number for the coresponding E3. ( If the score greater than 1, we let it be 1. We define results with “significance score”<0.001 as high confidence interaction, 0.001<“significance score”<0.01 as middle confidence interaction, 0.01<“significance score”< 0.05 as low confidence interaction and “significance score”> 0.05 as very low confidence interaction. The statistic table lists the result number belong to each confidence level.)

Figure 7. Confidence level
5.3 Filter
Filter is used to set the confidence range of the shown E3-substrate in the canvas.
For confidence mode, numbers on both sides of the slider are the minimum and maximum value of the confidence score range(Figure 6). You can use your mouse to drag the slider or modification the number in the textbox to change the range. The vertical line locates above the slider denotes the value of confidence scores for the shown E3-substrate interactions in the network.
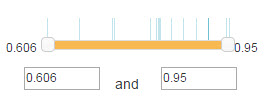
Figure 8. Confidence score filter
For evidence mode, you can filter the type of supporting evidence. Lines with different colors represent different supporting evidences. The number in the parentheses counts all the predicted E3-substrate interactions supported by this evidence. You can select/deselect the check box before each supporting evidences to show/hide E3-substrate interactions supporting by this evidence.

Figure 9. Supporting evidence filter
5.4 E3 hierarchical tree
680 E3 ligases are classified into different families. If you query a substrate, the predicted E3s and their position in the E3 family hierarchical tree will be presented. In this tree, texts in each circle (just like “F”, “D” and “SO”) represent the E3 family, the same as the text in node of Figure 5. The number in the bracket following each E3 family represents the number of corresponding predicted E3-substrate interaction and the number following each E3 gene symbol is the related confidence score. You can select/deselect one or more E3 types to show/hide E3-substrate interactions belong to this E3 type. The “download” button is used to download the prediction score of all the predicted results.
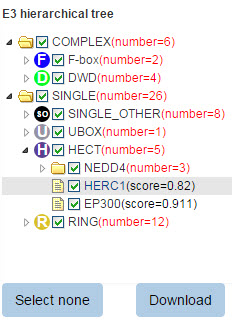
Figure 10. E3 hierarchical tree for substrate.
If you query an E3 ligase, the tree is look like this and just represents the E3 family classification.

Figure 11. E3 hierarchical tree for E3 ligases.
5.5 Network export
If you use Chrome browser, you can export the E3-substrate network graph by clicking the button “Export network” and the E3-substrate network graph will be saved as a PNG file. Otherwise, you can capture the E3-substrate network graph by other screen capture software.
5.6 Prediction summary
Because the E3-substrate network only show the top 20 predicted interactions. We construct a table to show all the predicted results for both confidence and evidence modes.
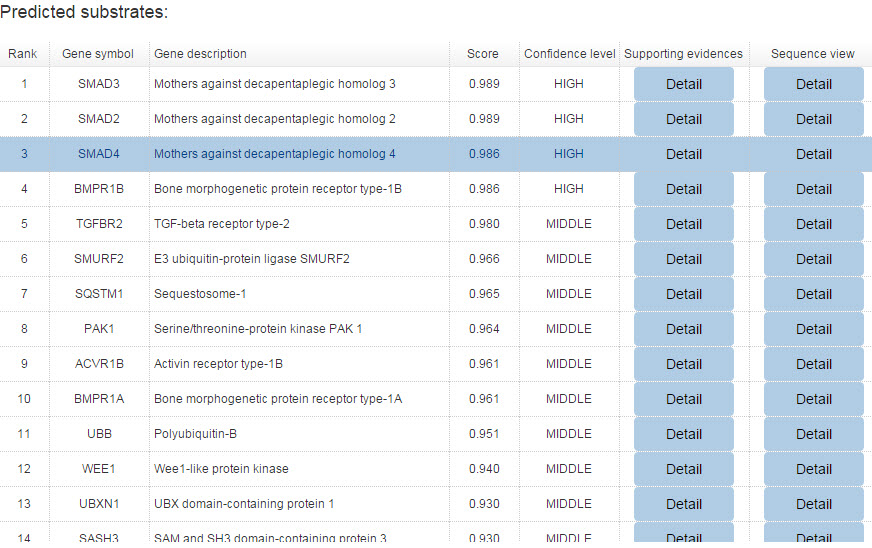
Figure 12. Summary table for confidence model
In Figure 13, for evidence mode, the gray levels of the dot reflect the predictive confidence from the related single evidence.
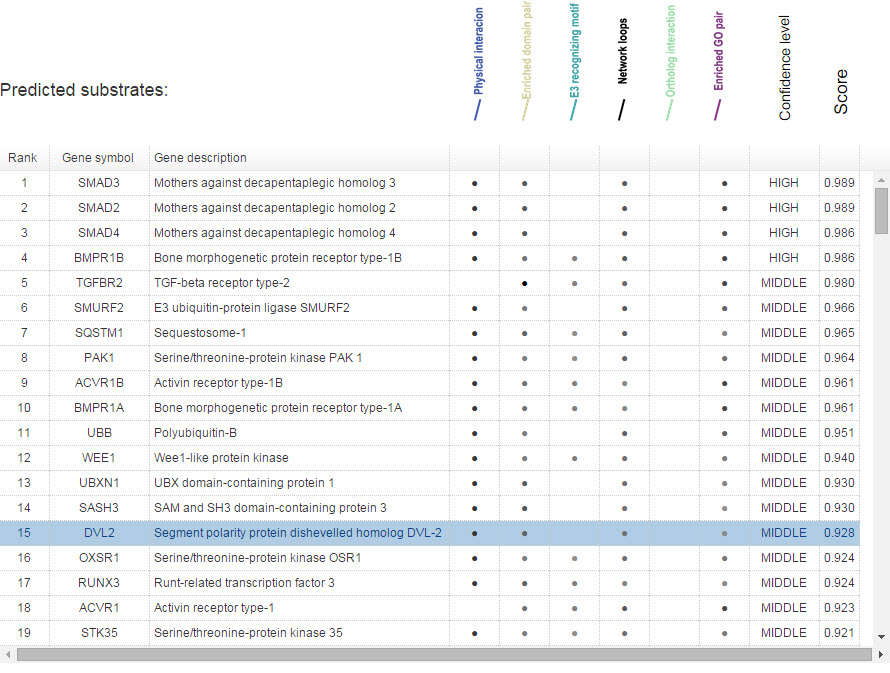
Figure 13. Summary table for evidence model
6 Supporting evidences for the predicted E3-substrate interaction
When you click the edge in the predicted E3-substrate network (link) or click the button in the supporting evidence row of summary table(link), a table will be presented to show the pages of supporting evidences for the E3-substrate interaction.
6.1 Brief information
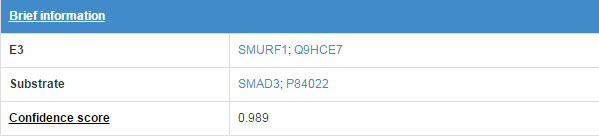
Figure 14. Brief information
*Detail of confidence score. (link)
6.2 Enriched domain pair
We predict the interaction domains between E3 and substrate. This table presents the interaction domain information.
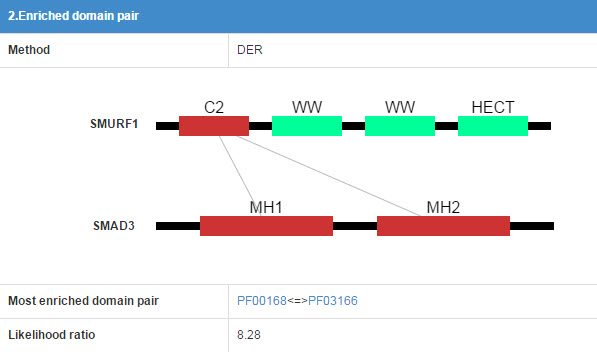
Figure 15 Table for enriched domain pair
Detail of the method of “Enrichment domain pair” and “DER”.
6.3 E3 recognizing motif
E3 may associate with the specific substrate by recognizing a short liner sequence motif, so we extract E3 recognizing motif for E3s. The first line of this table list the E3 recognizing motif extract method and you can click the method name to see detail of the method (part 10.7). The second line shows the Motif construction, “.” represent any amino acid. The third line is the likelihood ratio of this evidence.
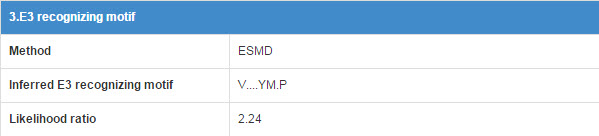
Figure 16. Table for E3 recognizing motif
6.4 Network loops
E3 and substrate will form some network topology mode in the physical interaction network. We count the numbers of three-interaction loops and four-interaction loops.
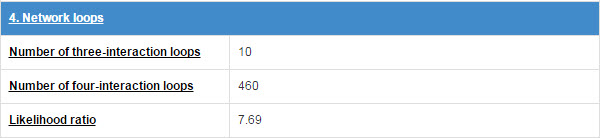
Figure 17. Table for network loops
6.5 Orthologous E3-substrate interaction
The orthologous E3-substrate interaction in mouse list in this table.
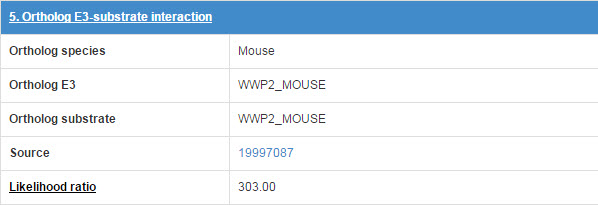
Figure 18 Table for Orthologous E3-substrate interaction
6.6 Enriched GO pair
E3 and substrate may have some functional association, so we calculate the GO annotation term pairs’ enrichment ratio between E3 and substrate.
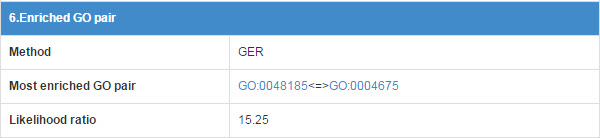
Figure 19. Table for enriched GO pair
*Detail of the method “Enrichment GO terms pair” of “DER”.
7 sequence view of the E3-substrate interaction
In the popped sequence view, the literature reported ubiquitination sites and predicted domains/motifs recognized by the corresponding E3 are marked by multiple signs.
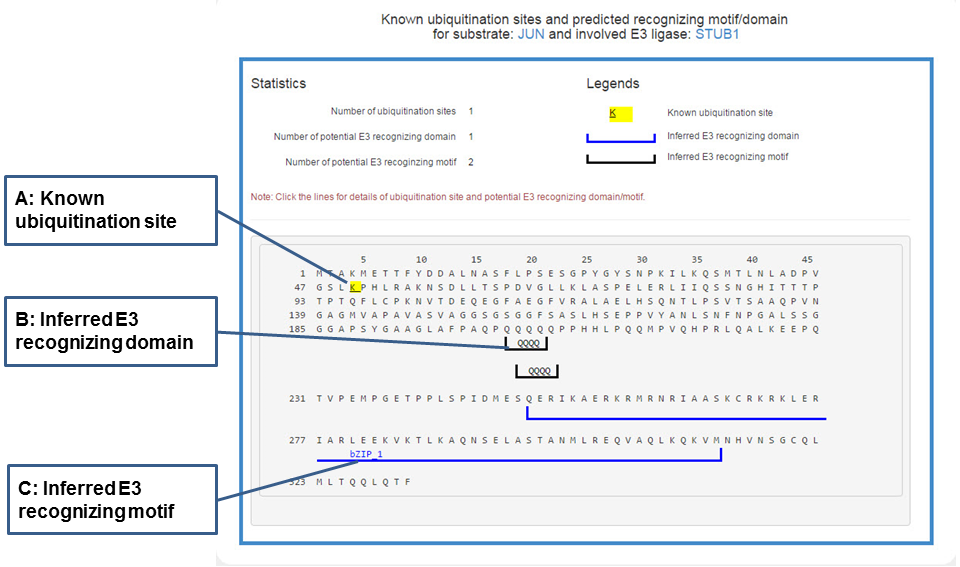
Figure 20. Sequence view of the E3-substrate interaction
The substrate’s sequence is shown in PRIDE format with multiple signs: black lines under the sequence denote the potential domain interacting with related E3, gray lines under the sequence mark the inferred E3 recognizing motif and the yellow background of character K means known ubiquitination site. By clicking all these signs you can get more information.
7.1 Info box for known ubiquitination site
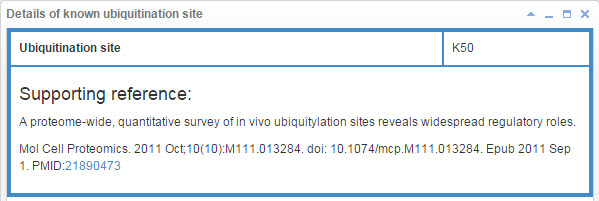
Figure 21. Info box for known ubiquitination site
7.2 Info box for inferred E3 recognizing domain
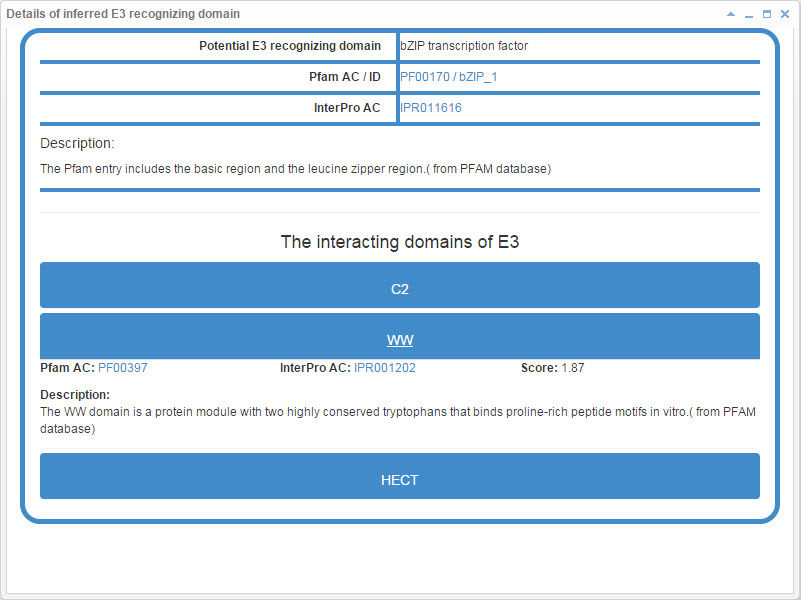
Figure 22. Info box for inferred E3 recognizing domain
7.3 Info box for inferred E3 recognizing motif
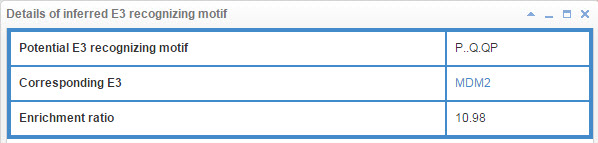
Figure 23. Info box for inferred E3 recognizing motif
8 Table view of the literature reported E3-substrate interaction
Figure 24. Table view of the literature reported E3-substrate interaction
9 Question and answer
If you have some questions, you can leave your questions. Clicking “Q&A” button on the navigation bar will jump to the question and answer page.
Figure 25. Question and answer
10 Nomenclature in UbiBrowser
10.1 Confidence score
The confidence score is computed as:
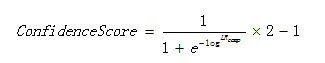
Equation 1
Where LR is the composed likelihood ratio.
10.2 Likelihood ratio
The Likelihood ratio of biological evidence f is the ratio of the probability of meeting condition f of interacting E3-substrate pair and non-interacting E3-substrate pair in the golden standard data sets. The Likelihood ratio is computed as:
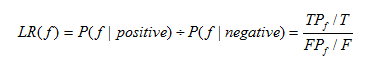
Equation 2
Where T and F are the number of all the true and false interactions respectively, TP and FP are the number of true and false interactions with the biological evidence f respectively.
10.3 Source database
The interaction databases recording this E3-substrate interactions.
10.4 Method
Distinct assessment method for one biological evidence.
10.5 DER
DER is shortness for Domain enrichment ratio.
10.6 Most enriched domain pair
The domain pair with the highest enrichment ratio.
10.7 Inferred E3 recognizing motif
Motif extracted from the golden positive datasets. the different characters represent different amino acids and "." represent any amino acids.
10.8 ESMD
Shortness for E3-substrate recognizing motif.
10.9 Number of three-interaction loops
Number of three-interaction loops that an E3-substrate interaction is involved in.
10.10 Number of four-interaction loops
Number of four-interaction loops that an E3-substrate interaction is involved in.
10.11 GER
GER is shortness for GO pair enrichment ratio
10.12 Most enriched GO pair
The GO term pair with the highest enrichment ratio.
11 Methods
11.1 Golden standard datasets
To evaluate each biological evidence, the golden standard positive and negative data sets are constructed. 1295 E3-substrate interactions were manually extracted from the literature downloaded from PubMed. These data were used as golden standard positive (GSP) dataset. The GSN (golden standard negative) dataset was defined as the randomly combination of human E3s and other human proteins, removing the overlap with GSP. Human E3 data was collected from UUCD (version 1.0).
11.2 Model organism E3-substrate interaction
366 pairs of mouse E3-substrate interactions were collected from E3net database. Pairwise ortholog map files were downloaded from the Inparanoid6 database (http://inparanoid.cgb.ki.se, Released 8.0), Human e3-substrate interactions were predicted by mapping mouse E3-substrate interactions to human orthologs using the Inparanoid database. Of 1295 ESIs in GSP, we predicted 301(23.2%) E3-substrate interactions while none in GSN was predicted.
11.3 E3-substrate enriched domain pairs
Considering that E3-substrate interactions are mediated by the interacting protein domains, we think that novel E3-substrate interactions may be predicted by identifying pairs of domains enriched among known E3-substrate interactions. Protein domain and family assignments were downloaded from Pfam (Released version 27). In total, 45019 assignments of 5487 protein domains and families to one or more of 18312 proteins were queried. Domain pair enrichment was assessed with the domain enrichment ratio (DER), which is calculated as the probability (Pr) of observing a pair of domain in a set of known E3-substrate interactions divided by the product of probabilities of observing each domain pair independently:
DER=Pr(d_e3:d_sub |GSP)÷(Pr(d_e3 |GSP)×Pr(d_sub |GSP)) (1)
where d_e3 is domain of E3 and d_sub is domain of substrate. d_e3:d_sub is a E3-substrate interaction in which E3 has d_e3 and substrate has d_sub, and GSP is a gold standard positive set of known E3-substrate interaction.
We used two thirds of the GSP to define enriched domain pairs and the remaining third to calculate the likelihood ratios. We repeated this process three times and combined these results. We found that the degree of domain enrichment in two thirds of GSP is strongly associated with the likelihood ratio calculated by the remaining third
11.4 GO term enriched pairs
An E3 and a protein that function in the same biological process, location in the same subcellular should be more likely to interact. To test this suppose, we calculated the GO term enrichment ratio (GER). The calculation is similar to E3 substrate enrichment ratio. GO annotation data was downloaded from Gene Ontology Consortium (http://geneontology.org Released November 27, 2014 ). Finally we found the degree of GO term enrichment is also strongly associated with the likelihood ratio.
11.5 Network topology structure
To identify the E3-substrate interactions with certain network topology, we combine the query interaction with the HPRD protein interaction data to generate an integrated network for explanatory variables correlating with their confidences. We define N3 and N4 as the number of the three- and four-interaction loops.
11.6 E3 substrate recognizing motif
E3 may associate with the specific substrate by recognizing a short liner sequence motif.
For each E3 in GSP, we predict its substrate recognizing motif based on two parallel sequence data sets: one is the sequence data of its substrates in GSP (target dataset) to build the motif, and the other is that of all the proteins interact with this E3 in GSP (background dataset) for background probability calculations. The reference sequence of human proteome was downloaded from Uniprot (version: May 2013) and the protein interaction datasets from HPRD.
To build the motif from a set of protein sequences, we in turn define the center amino acid from 20 kinds of amino acids.The first step is to convert both the target and background datasets into position-weight matrices (target matrix and background matrix) of equal dimensions where each matrix contains information on the frequency of all residues at the six positions upstream and downstream of the center amino acid. Using the information encoded in these two matrices, a third matrix, the hyper geometric probability matrix, is created. Specifically, this matrix contains the probability of observing s or more occurrences of residue x at position j (taken from the target matrix), given a background probability P for residue x at position j (taken from the background matrix).
The second motif-building step of the algorithm is a greedy recursive search of the sequence space to identify highly correlated residue/position pairs with the lowest P values. Each recursive iteration identifies the most statistically significant residue/position pair meeting a defined hyper geometric probability threshold (in this study taken as P < 10−3) and occurrence threshold (which represents the minimal number of sequences in the target data set needed to match the residue/ position pair). When such a pair is found, the sequence spaces of the target and background matrices are reduced by retaining only those sequences containing the selected residue/position pair, and a new hypergeometric probability matrix is calculated. This recursive pruning procedure is repeated until no more statistically significant residue/position pairs that meet the occurrence threshold are detected. At this point the motif is identified by the tally of residue/position pairs selected during this step and the its confidence score is defined as the sum of -log(P).The next step of the algorithm involves set reduction of the target and background data sets by removing all of those sequences that match the motif identified in the motif-building step. The purpose of this step is to remove the effects of those peptides with identified motifs from confounding the search for other significant motifs. Thus, performing the sequential loop of motif building is followed by set reduction results in a decomposition of the target sequence database into a list of significant motifs. The algorithm is complete when the motif-building step fails to identify any significant residue/position pairs. Finally, we assign another center amino acids and repeat the 3 steps above until all 20 types of amino acids are used as the center amino.